



Background
Dilated cardiomyopathy (DCM) is a clinical condition in which left ventricle (LV) dilation and subsequent impairment of its systolic function is caused by primary myocardial dysfunction [1]. The potential causes of LV dilation are vast and include myocardial destruction in the course of coronary heart diseases, toxic, infectious, or metabolic reasons [1 ESC]. Approximately 20-35% of cases with undetermined etiology are expected to have a familial form of DCM [2]. Familial DCM (FDCM) is in the vast majority a monogenic disorder transmitted in an autosomal dominant fashion. Most patients present between the ages of 20 and 60, but DCM can occur in children and older adults. The disorder is clinically very heterogeneous and may present as progressive left ventricular dysfunction with clinical signs of heart failure or malignant arrhythmias and sudden cardiac death (SCD). More than 30 genes encoding cytoskeletal or sarcomeric components have been associated with DCM [3].
A subset of patients presenting with DCM exhibit arrhythmias including paroxysmal or sustained atrial flutter or fibrillation, paroxysmal or sustained supraventricular arrhythmias,
ventricular tachycardia (VT) or ventricular fibrillation (VF) and/or conduction disorders. Mutations in the genes encoding lamin A and C nuclear envelope proteins (LMNA) and sodium channels (SCN5A) have been shown to be most prevalent in this group of DCM patients [4]. Additionally, presence of one of these mutations is related to an increase of SCD [5]. As a result, the latest Heart Rhythm Society (HRS) and the European Heart Rhythm
Association (EHRA) expert consensus recommends targeted screening for LMNA and SCN5A mutations in DCM patients with rhythm abnormalities while considering further management [6].
Case presentation
24 years old man with recurrent episodes of heart palpitation was admitted to our Centre for cardiological evaluation. He had a family history of dilated cardiomyopathy (DCM) and rhythm abnormalities. DCM was diagnosed in his mother, who died in the age of 45 and in his 10 years older brother, who remains under cardiological care. Moreover, his grandfather (father of the mother) died suddenly (SCD?) in the age of 36.
He has 5 siblings and none of them, apart from him and his older brother, has been diagnosed with any cardiac condition, so far. (Figure 1)
In the 24-hour ECG monitoring (Holter-ECG) carried out in February 2012 – Holter ECG: (no beta-blocker) 2149 ventricular extrasystoles (VES), 137 couplets, 14 sequences of max 5 beats were observed and in September 2012, 1793 VES together with 2 asymptomatic ventricular tachycardia (VT) with maximal duration of 3 seconds and max. 202 bpm and 7 episodes of idioventricular rhythm.
At admission, he was hemodynamically stable with no signs of pulmonary congestion or peripheral edema. He complained of only mild limitation of exercise tolerance, had no chest pain or vertigo. Biochemical analysis disclosed only mild dyslipidemia (total Cholesterol- 5,96mmol/L, LDL- 3,73 mmol/L, HDL- 0,97mmol/L, TG- 3,67mmol/L). Apart from that no abnormalities were detected. NT-proBNP level was 105 pg/ml and CK- 65 U/L. ECG revealed regular sinus rhythm, 66 beats per minute (bpm), no axis deviation, PQ- interval of 180ms, QTc- interval of 366ms, QRS complex of 80ms and horizontal ST segment depression of max 1 mm in II, III, aVF leads. Echocardiographic evaluation showed borderline left ventricle (LV) size, normal thickness of the LV walls, slightly decreased LV ejection fraction (LVEF) of 50%, regular LV filling pressure and no significant valvular abnormalities. (Table 1) Cardiac Magnetic Resonance (CMR) study disclosed enlarged LV of 236ml, thinning of the posterior and lateral LV wall with their hypokinesis, decreased global LV contractility, LVEF of 46%. (Table 2) No signs of right ventricle arrhythmogenic dysplasia (ARVD) or LV no-compaction (LVNC) has been revealed in the CMR. His exercise capacity was considered moderately impaired with 39,3 ml/kg/min of max VO2 consumption in cardiopulmonary exercise test (CPET).
He was put on beta-blocker (bisoprolol) 5mg once a day, propaphenone 150mg three times a day and ramipril 2,5mg once a day and potassium supplements. In Holter-ECG control study mean heart rate of 63 bpm, PQ-interval of 200ms and 2 single VES with no VT were observed.
He remains stable in the follow-up, hitherto.
Questions
1. Shall he be considered for an ICD implantation?
2. Is any genetic evaluation recommended?
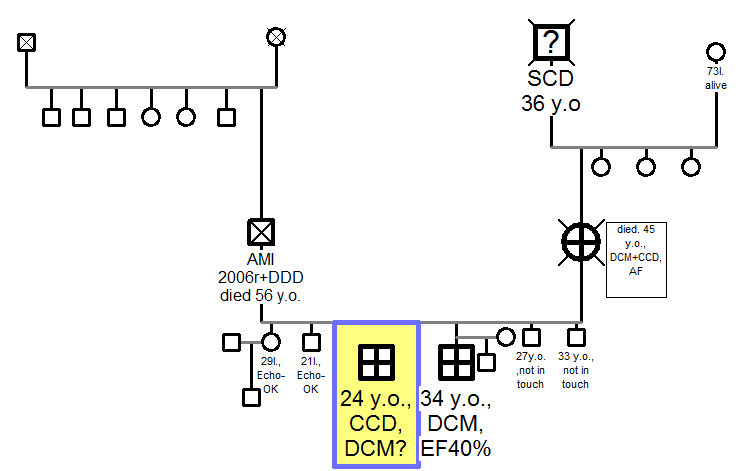
Figure 1. Family Pedigree. DCM: dilated cardiomyopathy; SCD: sudden cardiac death; AMI: acute myocardial infarction; DDD: post cardiac DDD stimulator implantation; AF: atrial fibrillation; CCD: cardiac conduction disorder; EF: ejection fraction.
|
Parameter |
Value |
|
LVDd |
52 mm |
|
IVSDd |
10 mm |
|
PWDd |
9 mm |
|
EF |
50% |
|
EDV |
136 ml |
|
RVDd (PLAX) |
26 mm |
|
TAPSE |
26 mm |
|
LADd |
29x36x44 mm / 13 cm2 |
|
RADd |
41×36 mm / 12 cm2 |
|
Mitral valve |
E- 0,74 m/s, E/A- 1,53, E’- 0,09 m/s, E/E’- 7 |
|
RVSP |
25 mmHg |
|
Table 1. Transthoracic cardiac echo study. Values. LVDd: left ventricle diastolic diameter; IVSDd: intraventricular septum diastolic diameter; PWDd: posterior wall diastolic diameter; EF: ejection fraction; EDV: end-diastolic volume; RVDd: right ventricle diastolic diameter; PLAX: parasternal long axis view; TAPSE: Tricuspid Annular Plane Systolic Excursion; LADd: left atrium diastolic diameters; RADd: right atrium diastolic diameters; RVSP: right ventricle systolic pressure. |
|
|
Parameter |
Value |
|
LVDd |
62 mm |
|
IVSDd |
9 mm |
|
PWDd |
5 mm |
|
EDV |
236 ml |
|
EF |
46% |
|
Mass |
160 g |
|
RVD |
23 mm |
|
LAD |
18 cm2 |
|
RAD |
21 cm2 |
|
Table 2. Cardiac Magnetic resonance. Values. LVDd: left ventricle diastolic diameter; IVSDd: intraventricular septum diastolic diameter; PWDd: posterior wall diastolic diameter; EF: ejection fraction; EDV: end-diastolic volume; RVD: right ventricle diastolic diameter; LAD: left atrium diastolic diameters; RAD: right atrium diastolic diameters; |
|
Current guidelines
• ICD therapy is indicated in patients with nonischemic DCM who have an LVEF ≤35% and who are in NYHA II or III (I;B), (ACC/AHA/HRS 2008 Guidelines for Device-Based Therapy of Cardiac Rhythm Abnormalities; Circulation 2008) + (ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012; EHJ 2012)
• ICD may be considered in patients with DCM with ejection fraction >35 percent with family history of sudden cardiac death OR with LMNA mutation, (Genetic Evaluation of Cardiomyopathy, A Heart Failure Society of America Practice Guideline; J Card Fail 2009)
• ICD in patients with DCM with syndromic disease (muscular dystrophy) or known arrhythmia and/or conduction system disease (LMNA or desmin [DES]) may be considered, (HRS/EHRA Expert Consensus Statement on the State of Genetic Testing for the Channelopathies and Cardiomyopathies; Europace 2011)
References
1. Elliott PM, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position
statement from the european society of cardiology working group on myocardial and pericardial diseases. Eur Heart J 2008;29:270–276
2. Hershberger RE, Lindenfeld J, Mestroni L, et al. Heart Failure Society of America. Genetic evaluation of cardiomyopathy: a Heart Failure Society of America practice guideline. J Card Fail 2009;15:83-97.
3. Rapezzi C, Arbustini E, Caforio ALP, et al. Diagnostic work-up in cardiomyopathies: bridging
the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2012, epub ehead.
4. Kushner JD, Nauman D, Burgess D, et al. Clinical characteristics of 304 kindreds evaluated for
familial dilated cardiomyopathy. J Cardiac Failure. 2006; 12:422–29.
5. Gollob MH, Blier L, Brugada R, et al. Society Position Statement Recommendations for the Use of Genetic Testing in the Clinical Evaluation of Inherited Cardiac Arrhythmias Associated with Sudden Cardiac Death: Canadian Cardiovascular Society/Canadian Heart Rhythm Society Joint Position Paper. Can J Cardiol 2011;27:232–245
6. Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA Expert Consensus Statement on the
State of Genetic Testing for the Channelopathies and Cardiomyopathies. Europace 2011;13:1077–1109
Expert’s comments:(Written authorization required from each expert)
1. Jacek Bednarek, MD, PhD
This very young patient currently has no classical indications for and ICD implantation in primary prevention. Genetic evaluation together with endomyocardial biopsy may be recommended in order to support decision making regarding further management. Re-considaration for an ICD implantation should be performed after receiving results of abovementioned tests.
2. Lidia Tomkiewicz- Pająk MD, PhD
Regarding his young age and no apparent clinical or echoradiographical features od DCM he should be regularly followed-up with cardiac echo or magnetic resonance evaluation, cardio-pulmonary exercise testing, Holter ECG. Genetic evaluation is recommended prior the decision of and ICD implantation as an primary prevention.
3. Piotr Podolec MD, PhD
Patient should be followed-up regularly (6-12 months) with evaluation of any dilated cardiomyopathy features in cardiac magnetic resonance or echocardiographic studies. Since there has not been history of myocarditis no biopsy should be now recommended. Instead, genetic evaluation may add to consideration of further management.
Expert’s conclusions:
Patients remains in close follow-up group. He should have cardiac echo study or cardiac magnetic resonance performed every 6- 12 months together with Holter Ecg and cardio-pulmonary tests. Genetic evaluation is recommended. Re-consideration for an ICD implantation in primary sudden cardiac death prevention should be carried out with the results of abovementioned tests.
Authors:
Jakub Stępniewski MD1, Paweł Rubiś MD, PhD1, Agata Leśniak-Sobelga MD PhD1, Magdalena Kostkiewicz MD, PhD.1
Experts:
Jacek Bednarek2, MD, PhD, Lidia Tomkiewicz- Pająk MD, PhD1, Piotr Podolec MD, PhD1
1Department of Cardiac and Vascular Disease in John Paul II Hospital, Institute of Cardiology, Faculty of Medicine, Jagiellonian University, Krakow, Poland.
2Department of Electrocardiology in John Paul II Hospital, Institute of Cardiology, Faculty of Medicine, Jagiellonian University, Krakow, Poland.
